异相芬顿反应(Heterogeneous Fenton Reaction)因其可避免传统Fenton体系的铁泥、低pH等限制,受到广泛关注。然后,大部分研究主要集中于如何构建更高效的异相Fenton体系及其自由基作用机理,作者针对不同吸附性污染物在异相Fenton体系下可降解性差异,构建了简单的水铁矿-双氧水(H2O2)体系,结合表面自由基的扩散构建多个模型,提出界面层概念,更新了对异相芬顿的认识。基于此,澳大利亚新南威尔士大学土木与环境工程学院T. David Waite院士课题组于2021年在Environmental Science & Technology上发表了题为“Heterogeneous Fenton Chemistry Revisited: Mechanistic Insights from Ferrihydrite-Mediated Oxidation of Formate and Oxalate”的研究论文。
受长江流域环境水科学研究公众号邀请,中国地质大学(武汉)袁松虎教授课题组博士生张耀强对本文进行了解读
第一部分:内容解读
摘要:非均相Fenton反应已有广泛研究,但对该工艺的某些机理认识仍不全面。本研究中,我们量化了pH 4.0下,甲酸盐和H2O2在新合成水铁矿表面的吸附和分解,并借助动力学和反应迁移模型,为水铁矿介导下的非均相Fenton反应机理提供了新的见解。结果表明,H2O2和甲酸盐的分解受表面引发反应的控制。被吸附的甲酸盐占据了可与H2O2反应的表面位点,因此阻碍表面Fenton反应,尽管H2O2在表面的积累可以忽略不计。甲醇淬灭HO•对甲酸盐氧化的影响很小,同时,相较于完全吸附的草酸盐较少氧化,部分吸附的甲酸盐氧化显著,这表明氧化主要发生在固-液边界层,而不是水相或表面。动力学和反应输运模型结果表明这可能是由于表面生成的羟基自由基(HO•)扩散,而非表面Fe(II)扩散到边界层导致。这些新发现对我们理解更复杂的目标有机污染物去除,以及更有效的非均相Fenton技术设计至关重要。
研究背景
异相Fenton技术中,固相催化剂表面活性位点介导H2O2的分解和HO•的产生,当前研究主要集中如何构建更高效的异相Fenton体系及其自由基机理过程。直觉上,被固相催化剂吸附的污染物更容易接触到在表面或近表面部位形成的氧化剂,因而更容易降解。部分研究确实将此作为其污染物高效降解的原因之一,认为污染物降解发生在表面区域(Surface)。同时,一些研究中发现,被吸附只固相表面的有机物并没有被氧化,由此认为表面产生的HO•主要作用于水相区域(Liquid bulk)。
作者从污染物与自由基反应位置的角度重新审视(Revisited)异相Fenton反应,将HO•的扩散考虑至体系中,提出污染物降解可能主要在固-液界面层的新观点。通过水铁矿介导下H2O2分解产HO•对甲酸(部分吸附)/草酸(完全吸附)的降解,验证其猜想,并一进步通过动力学模型定量刻画这一过程。
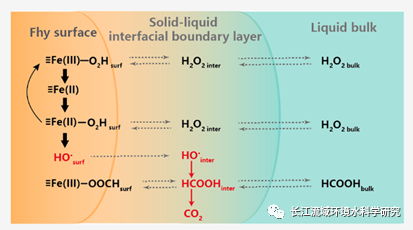
图片来源:https://pubs.acs.org/doi/10.1021/acs.est.1c00284。
结果部分(假说论证)
本论文的实验体系设计非常简单但也颇为巧妙。水铁矿介导下的双氧水异相Fenton促进甲酸/草酸的分解,通过直接过滤和加酸溶解水铁矿释放吸附态物质两种取样测试方式,定量氧化和吸附对甲酸/草酸以及H2O2的相对贡献。论文主要逻辑为,通过批实验提供确切的实验现象规律和反应速率,通过动力学模型和简单的反应迁移模型对其机理猜想进行验证。
在批实验中,通过改变H2O2、甲酸/草酸、水铁矿的初始浓度,测试不同条件下双氧水和有机物的分解速率,以及甲醇淬灭实验。论文获得了以下实验现象层次结论(图比较多,就简略总结如下,具体论证细节可参考原文):(1)反应过程中,均相的溶解态Fe(III)含量极低,使得双氧水和甲酸的均相Fenton分解影响可忽略;(2)甲醇(水相HO•淬灭剂,电中性,不容易被固相催化剂吸附)淬灭对甲酸的氧化影响较小,表明甲酸氧化过程发生在界面或者边界层(HO•扩散迁移距离有限);(3)完全吸附的草酸几乎无氧化,部分吸附的草酸显著氧化,表明氧化过程发生于边界层而非界面区域;(4)双氧水分解速率与固相位点数量(根据文献中单位水铁矿量反应位点含量以及有机质吸附量估算固相位点数量)成正相关,表明吸附到固相表面的甲酸和草酸能够占据可与双氧水反应的表面活性位点,降低双氧水分解速率;(5)H2O2分解速率与其初始浓度正比,吸附态H2O2测试不到,且不同初始浓度H2O2对甲酸盐的吸附几乎不影响,表明双氧水几乎不占据反应位点。
上述批实验已经相对完整,作者进一步通过动力学模型对于反应现象进行机理解释。将上述异相Fenton体系中所有可能发生的反应均纳入一个动力学模型中,如下图所示,主要的反应过程包括四个部分:(1)均相反应过程;(2)甲酸、双氧水、中间体自由基、HO•的扩散;(3)表面Fenton反应;(4)界面层Fenton反应;

图片来源:https://pubs.acs.org/doi/10.1021/acs.est.1c00284。
上述反应模型具有大量的未知反应速率,如果全部通过耦合的方式,则难以区分具体的机理认识。由此,将机理过程进行分类,通过设置极限情况,观察最终结果是否与实验现象相符,来获取可能的机理假设组合。如下图所示,总共包括三大类合计8种可能的机理组合。(1)基本假设:a. HO•从界面产生并扩散至边界层;b.Fe(II)从界面扩散至边界层并与此处的双氧水反应在边界层产生HO•;用于解释实验中,有机质氧化发生于边界层的现象(2)a. H2O2从水相至界面的扩散是速率限制步骤,b.并非速率限制因素,仅因为H2O2从水相至界面的扩散速率比界面至水相扩散慢;用于解释实验中H2O2不占据反应位点,且不吸附的结果;(3)中间体自由基的扩散:a.链式反应的中间自由基迅速从表面迁移,不贡献表面Fe(III)的还原,b.这部分中间体自由基对表面Fe(III)的还原有贡献。
通过模拟结果参数的耦合,仅能确定双氧水的扩散不是限速步骤这一个猜想。对于是HO•还是Fe(II)的扩散,无法确定。于此同时,发现假设3对实验结果影响较少,因此对此部分采用最佳耦合的方式确定参数。
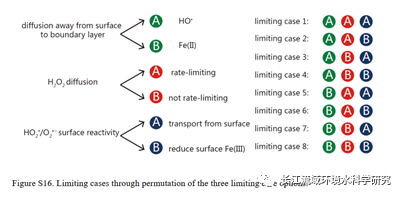
图片来源:https://pubs.acs.org/doi/10.1021/acs.est.1c00284。
作者进一步构建了一个简单的自由基反应迁移模型,来确认有机质在边界层氧化源于界面区域的HO•扩散还是Fe(II)的扩散至边界层。反应迁移模型表明,如果是Fe(II)的扩散,这Fe(II)将会不可避免扩散至水相中,在水相中同样产生HO•,则模拟结果将与甲醇淬灭实验结果相违背,因此确定氧化反应主要源于HO•的扩散。此外,作者还对模型进行了主成分敏感性分析,以及通过动力学模型对异相Fenton中有机质非自由基途径降解和有机质氧化发生在表面两种可能性进行了模拟,排除了上述可能性或者影响不显著。
讨论部分(成果意义):
论文揭示在水铁矿介导H2O2分解的异相Fenton体系中,甲酸的分解是在界面层中发生,且贡献主要源于表面产生并扩散至边界层的HO•。同时,有机质吸附在固相催化剂表面,会占据能够与双氧水反应的活性位点,降低双氧水分解速率。此认识对原位化学氧化过程中,有机污染物在地下环境中的氧化降解具有关键意义,如吸附性污染物可能由于(1)缺乏与边界层暴露,(2)占据H2O2反应位点,导致氧化效率降低。基于上述认识,为构建更高效的异相Fenton技术提供了理论指导。此外,在对异相Fenton体系进行机理探索时,如使用表面淬灭剂时,需考虑表面淬灭剂占据活性位点导致氧化效率降低带来的影响。最后,论文中构建的动力学模型,可以用来评估异相Fenton技术的修复效率。
第二部分:贡献解读
背景问题:异相Fenton反应是一个被广泛关注和研究的体系,研究主要集中如何构建更高效的异相Fenton体系及其自由基机理过程。作者关注到,对于不同吸附性的污染物种类,在异相Fenton体系中,其效率存在显著的差异,对反应位置的解释也不同。这也成为本论文的切入点,围绕这个关键科学问题,简化相应的体系设计,构建批实验和模型论证其观点。
核心发现:提出边界层的概念,从空间角度更新了对异相Fenton反应中有机质氧化反应位点的认识。对于认识,基于注入双氧水、过硫酸盐等氧化剂的原位化学氧化修复过程中,有机物污染的降解,及后续如何优化原位氧化技术设计和调控具有理论指导意义。
本文由中国地质大学(武汉)生物地质与环境地质国家重点实验室袁松虎教授课题组博士生张耀强解读。受作者能力所限,本文难免有不当之处,敬请各位读者谅解。如疑义、建议或其他方面的学术交流,请于袁松虎教授联系,邮箱yuansonghu622@cug.edu.cn.
